Quick start¶
We will proceed directly to a run of the box-model system, to show how
chemical schemes are normally compiled into CM_ and CMX_ files, and used
in box-model simulations. This is actually the normal and recommended way to
prepare files for the EMEP model, but also provides a good environment for
comparing chemical mechanisms.
Step 1: initial setup¶
If not run previously, some preliminary steps are needed to set up a working directory. From the GenChem/box directory, do:
cd somepath/GenChem-xxx/box
scripts/box_setup.sh tmp_work
The name _tmp_work is just an example - anything can be used.
Step 2: do.testChems¶
At this stage, one can try compiling a chemical scheme. With the example of EmChem19a, and now from our tmp_work directory, try:
cd tmp_work
./do.testChems EmChem19a
This script will run GenChem.py on the EmChem19a scheme (also adding a few extra reactions from helper BoxAero, BoxBVOCemis and BoxDep mechanisms), run “make”, and then run the resulting box-model code. Results will appear in one log-file (e.g. RES.EmChem19a), and as comma-separated results in the Output directory (set in do.testChems): OUTPUTS_TEST/boxEmChem19a.csv. This file is readable with e.g. libreoffice. Plot scripts are also available (see next section), for easy visualisation and comparison of these csv results.
The CM_ and CMX_ fortran files produced by this process are saved
in directories, e.g. here in ZCMBOX_EmChem19a. These files could
be used in the EMEP model if wanted, but usually the more complex script
emep_setup.py (described below) is used for that. (Hence we reserve
the prefix ZCMBOX for files created by do.testChems and ZCM for those
created with emep_setup.py, see below.)
Now, if one wants to compare several schemes, one can do e.g.:
./do.testChems EmChem19a CRIv2R5Em MCMv3.3Em
This would produce 3 output .csv files, which again are easily plotted against each other.
Technical comments:
- do.testChems is just a simple wrapper, which cleans up files, runs another script (do.GenChem), compiles, and runs the box model, boxChem.
- MCM is a very large scheme and this can take a while, or stress your PC’s memory! Try with the smaller schemes first.
2a. Plotting?¶
If one has run say 3 chemical schemes using Step 2 above, the results are easily plotted from the box/tmp_work/OUTPUT_TEST directory:
../../scripts/boxplots.py -h for help!
../../scripts/boxplots.py -v O3 -i boxEmChem19a.csv boxChem1.csv boxChem2.csv -p
Using ‘ALL’ or ‘DEF’ with -v results in all or many common species being plotted at once (-p is assumed in this case). For example, here we can see a comparison of three schemes produced with this script:
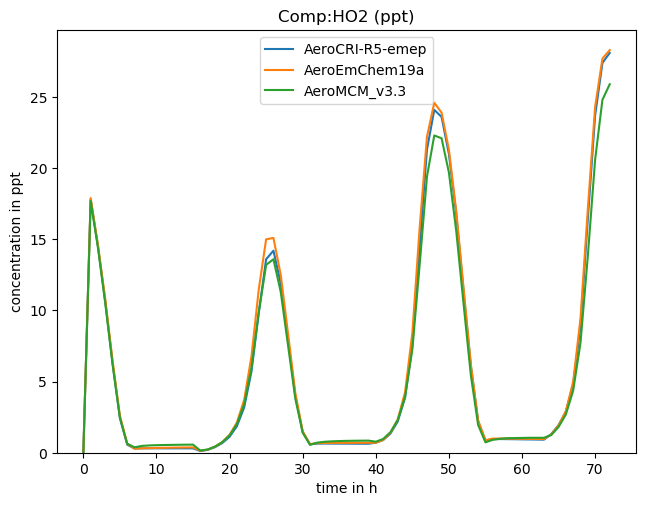
Another crude+helpful script just grabs the concentrations:
../../scripts/getboxconcs.py O3 boxEmChem19a.csv
which results in ResConcs_boxEmChem19a_O3_ppb.txt
2b. Box-config¶
The script do.testChems above compiles the executable boxChem for each mechanism in turn, and by default runs this using some settings from the default config_box.nml file. This file contains a number of important settings which by deault run a 24-hour simulation (starting at 12:00 GMT), with set emissions, temperature of 298.15 K, mixing height of 1000 m, and some boundary conditions. Default outputs are also given.
The user can of course change these settings (do this in your working directory, not in src). We explain some the key variables and choices here, and further information can be found in Simpson et al 2020.
Note these config files follow fortran namelist conventions. Text following an exclamation mark (!) is ignored.
Geographical location¶
lat = 45.05, ! degrees N
lon = 15.06, ! degrees E
Emissions¶
use_emis = T, ! use emissions at all?
! directory with emissplit files:
emissplit_dir = 'emissplit_run/'
emis_kgm2day = 'nox', 18.3, ! NOx, kg/m2/day, as in MCM/CRI tests
'voc', 15.4 ! NMVOC
!emis_kgm2day = 'nox', 180.3, ! NOx, kg/m2/day, as in MCM/CRI tests
! 'voc', 150.4 ! NMVOC
! BVOC emissions are set in chem/extra_mechanisms/BoxBVOCemis, where
! also a factor SUN is given for light-dependent emissions. These BVOC
! emissions can be adjusted with the factors below.
fIso = 1.0, ! isoprene
fMTL = 0.0, ! monoterpenes from light-dependent emissions
fMTP = 0.0, ! monoterpenes from pool (Temp.)-dependent emissions
fSQT = 0.0, ! sesqui-terpenes
Outputs¶
! Can say just e.g. 'O3', to reduce size of outputs,
! but in general usage 'all' is normally best.
OutSpecs_list =
'all', 'ppb' ! Will switch to ug for OM
! Output Groups
! --------------
OutGroups_list =
'NOX', 'ppb',
Debug¶
Some flags produce more output. More documentation to be added later.
! -----
! For testing, one can assign all VOC to one species. Do that here:
! dbgVOC = 'NODEBUG',
! dbgVOC = 'C2H4',
debug%Emis = 0
! debug%VOC = 'C2H4'
debug%Spec = 'NONE', !'C2H4'
debug%SOA = 0
debug%PM = .false.
debug%Chem = .false.
Step 3: emep_setup.py¶
The do.testChems script described above is best for quickly testing and comparing different mechanisms. Usually these comparisons only involve gas-phase mechanisms such as EmChem19a or MCMv3.3Em. However, the EMEP model usually requires a host of extra species and reactions to accommodate sea-salt, dust, organic aerosols, and pollen. It also requires files to specify how emissions and boundary conditions should be distributed among specific species, e.g. how a VOC emission should be split into C2H6, C2H4, nC4H10 etc.
In fact, for the EMEP model, GenChem produces many files which are copied into ZCM_XXX directories for the scheme XXX you wish to use:
$ls -x ZCM_EmChem19a/
CM_ChemDims_mod.f90 CM_ChemGroups_mod.f90 CM_ChemRates_mod.f90 CM_ChemSpecs_mod.f90 CM_DryDep.inc CM_EmisFile.inc CM_emislist.csv CM_EmisSpecs.inc CM_Reactions1.inc CM_Reactions2.inc CM_Reactions.log CM_WetDep.inc CMX_BiomassBurningMapping_FINNv1.5.txt CMX_BiomassBurningMapping_GFASv1.txt CMX_BoundaryConditions.txt config_box.nml run_emislist/ (with emislist_defaults_sox.csv etc..)
The recommended way to get this directory is to use the script emep_setup.py from your temporary work directory within the box system. So, from e.g. box/tmp_work, do:
./emep_setup.py EmChem19a
or just:
./emep_setup.py
and this will provide a list of options.
You can edit the emep_setup.py scripts, maybe renaming it as my_setup.py directory. If selecting from the provided base_mechanisms and extra_mechanisms you only need to extend the possible command lines as provided by the cmdx dictionary:
cmdx['EmChem19a-vbs'] ='-b EmChem19a -e PM_VBS_EmChem19 '+common_IsoMT1
cmdx['CRIv2R5Em-M19'] ='-b CRIv2R5Em -e PM_JPAC_MT3 PM_Hodzic_Aromatics BVOC_XTERP_CRI'+common_IsoMT3'
The ‘-b’ argument gives the base mechanism, and then you can have any number of compatable extra mechanisms (-e argument).
(There are many possible combinations of packages - see Simpson et al 2020
and the emep_setup.py code for many examples.)
Any keys from cmdx can be used by emep_setup.py. For example, if the user builds a new base scheme usersChem and some OA scheme, usersSOA, then emep_setup.py can be edited to add these as a new option:
cmdx['usersChem'] ='-b usersChem -e usersSOA'+common
you could do:
do.testChems usersChem # GOOD TO CHECK FIRST
emep_setup.py usersChem # Creates ZCM_usersChem
UPDATE 2022-06: Choice of GNFR_CAMS or SNAP¶
GenChem was originally developed when the EMEP CTM used the so-called SNAP emission sector system, which has 11 source categories. The EMEP model currently uses a 19-sector system we refer to as GNFR_CAMS. To produce EMEP-ready files with GenChem one now needs to specify “-g” in the call to emep_setup.py, e.g.
./emep_setup.py EmChem19a-vbs -g
Omission of this -g flag simply gives the 11-sector SNAP splits, though this option is now deprecated.
Step 4: Use for EMEP CTM¶
After emep_setup.py has successfully run, the ZCM_ directory produced contains all the files needed to run the EMEP CTM. The CM_ and CMX_ files can be copied directly to the CTM’s source directory, and the EMEP model compiled as normal (make clean, make). The emissplit_run files need to be sent to a location specified by the user (via the EMEP CTMs’ emep_config.nml namelist).